Author: Hongyan Hao
Editor: Keith Fraga
It has been more than 150 years since Gregor Mendel’s pea plant experiment demonstrated that ‘invisible factors’ determine the traits of organisms. It is doubtful that it would cross his mind that one day people could directly make changes to the “invisible factors”, which we call genes today, and alter these traits. Since the first transgenic Escherichia coli obtained antibiotic resistance in 1970s, tools have been developed to modify the genes of microbes, cell lines, plants, animals, and even human patients.
The principle of gene editing is based on the observation that cells can repair double strand breaks (DSB) through either the error prone pathway called non-homologous end joining (NHEJ) which sticks the two ends together or homologous recombination (HR) pathway that precisely repair the break using a template. Thus, the key to edit genes is to induce a site-specific DSB in the DNA sequence and allow the cell’s HR machinery to introduce the edit.
Standing out as a novel tool, the Clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated protein 9 (Cas9) system was soon appreciated and widely used by biologists for gene editing since its discovery. Originally found in bacteria, CRISPR arrays contain short repeated DNA sequences separated by unique spacers acquired from phage DNA (protospacer) upon previous infection. When the same phage DNA is detected, the CRISPR array processes the corresponding CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA) that form a duplex to bring the nuclease Cas9 to the matching external protospacer DNA and destroy it. The phage DNA contains a 5’-NGG-3’ protospacer-adjacent motif (PAM) which is not present in the CRISPR array, so that the system can distinguish self-sequence and the phage DNA .
Given the ability to specifically cut the external phage DNA upon reinfection, the bacterial CRISPR/Cas9 system was soon adapted and developed as a tool for gene editing. CRISPR/Cas9 uses a single nuclease Cas9 and two RNA molecules to perform site-directed DNA editing. The crRNA identifies target DNA and the tracrRNA glues the crRNA to Cas9 to direct the protein to target DNA. Notably, a single guide RNA (gRNA) combines crRNA and tracrRNA into a single RNA molecule also has the full gene-editing activity in vivo and in vitro. Thus, a designed gRNA combines with Cas9 protein makes a versatile, cheap, easy to use, and efficient gene-editing tool.
Detailed structural biology studies has helped understand the complex mechanisms to this critical aspects of CRISPR/Cas9 action (reviewed by Jiang et. al. 2017). How does the Cas9/gRNA complex search the genome for the target? How is the nuclease activity of Cas9 only simulated at matching sequences? How prevalent are off-target effects, and how can the practitioner avoid these effects?
How does the Cas9/gRNA complex search the genome for the target?
Cas9 is structurally organized into two distinct lobes, the target DNA recognition lobe (REC) that and the nuclease (NUC) lobe. The recognition lobe is made up of three alpha-helical domains, and the NUC lobe contains HNH-like and RuvC-like nuclease domains that create a break on each strand, as well as the C-terminal domain (CTD), which contains the PAM recognition site. The two lobes are connected by an arginine-rich linker (figure 1). Three major regulatory steps occur to achieve the efficient yet specific cut.
- First, the gRNA binds Cas9 which leads to a large conformational change to activate Cas9 (Figure 1).
- Second, the Cas9-gRNA complex binds to the target DNA PAM. PAM binding melts the adjacent DNA and plays an important role in crRNA/target-DNA strand hybrid formation (Figure 2).
- Third, the perfect match of crRNA and the target strand induces another conformational change which activate the nuclease activity to make the double strand break (Figure 3).
gRNA loading is the key for CRISPR to work. Without gRNA binding, the Cas9 protein is inactive and binds DNA weakly and nonspecifically. Structural studies also revealed there is a large conformational change in Cas9 where Helix-III (Hel-III) in the REC domain moves towards the HNH nuclease domain upon guide RNA loading, illustrated in the Figure 1 cartoon.
Cas9/gRNA complex recognizes a 20-nucleotide (nt) complementary DNA sequence adjacent to the 5’-NGG-3’ PAM. With the conformational change, the 5’-NGG-3’ PAM recognition sites in the CTD domains are also repositioned to be able to form base-specific hydrogen bonds with the conserved GG (Figure 1). Moreover, the ribose-phosphate backbone of the guide RNA contacts with Cas9 and the 3’ 10-nt crRNA sequence (seed sequence) is prepositioned in a target binding favorable form. Now, the Cas9/gRNA complex is ready for target search.
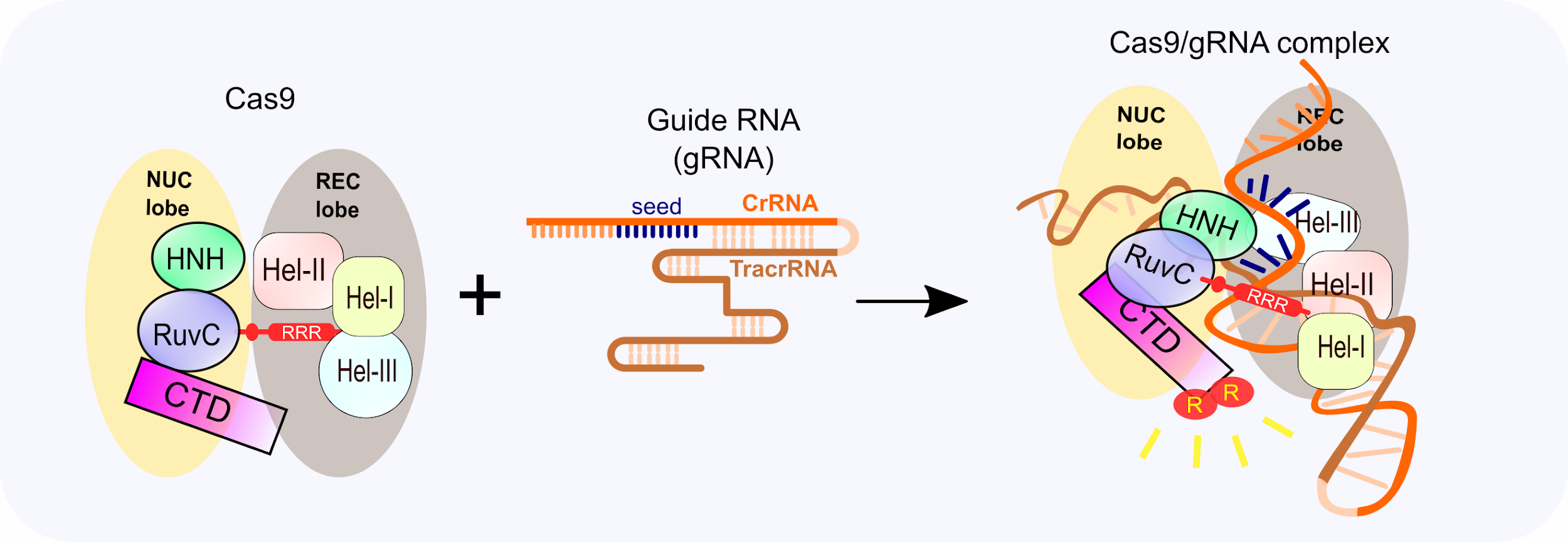 Figure 1: Cas9 protein contains the nuclease (NUC) lobe and the recognition lobe (REC) that are connected by an arginine-rich linker. Two arginines in the CTD (C-terminal domain)are exposed upon guide RNA(gRNA) loading, which will search for the target DNA PAM site and bind to it.
Figure 1: Cas9 protein contains the nuclease (NUC) lobe and the recognition lobe (REC) that are connected by an arginine-rich linker. Two arginines in the CTD (C-terminal domain)are exposed upon guide RNA(gRNA) loading, which will search for the target DNA PAM site and bind to it.
Single molecule studies have revealed that Cas9/gRNA searches its target in a three-dimensional way rather than sliding on the DNA sequence. PAM probing is the key step to direct the Cas9 to potential targets. The base-specific hydrogen bonds between the GG and the R1333 and R1335 in the CTD domain are made in the major groove of DNA, which confers higher sequence specificity. A single mutation in the PAM site to 5’-NCG-3’ abolishes the double strand break created by CRISPR/Cas9. Interestingly, Cas9 with engineered (T1337R) PAM recognition site engages a fourth guanine could recognize 5’-NGNG-3’ to introduce the DSB, further supports the importance of PAM recognition by CRISPR/Cas9.
How is the nuclease activity of Cas9 only simulated at matching sequences?
PAM-binding destabilizes the DNA duplex and triggers the Watson-Crick base pair between the crRNA and the target DNA (Figure 2). Next a series of three dynamic conformational changes occur to favor the DNA/crRNA hybrid formation, which is the rate-limiting step for Cas9/gRNA to make the double strand break revealed by kinetic studies.
- First, the hydrogen-bond association of PAM with the CTD domain results in longer binding time between the gRNA and target DNA, which enables the following RNA/DNA hybrid formation.
- Second, the phosphate in the target strand adjacent to the 5’ end of the PAM undergoes an unfavorable kink turn, which is stabilized by a phosphate lock loop (K1107-S1109) in Cas9. This kink contributes to both DNA double helix unwinding and the crRNA/DNA duplex formation.
- Third, Cas9 CTD-domain makes Van der Waals interactions with the DNA phosphate backbone of the PAM-containing non-target strand. The nucleotide immediately upstream of PAM in the non-target strand stacks on the PAM duplex to stabilize it and the non-target strand kinks. The disordered non-target strand is then stabilized by the interaction between Cas9 and the -2 and -3 nucleotides. The kink in the non-target strand also helps to expose the two seed nucleotide strand to initiate RNA/DNA duplex formation. This is consistent with the observation that the mismatch of the two-crRNA nucleotide adjacent to PAM is intolerable while off-target DSB could occur in the DNA sequence homology to the seed region.
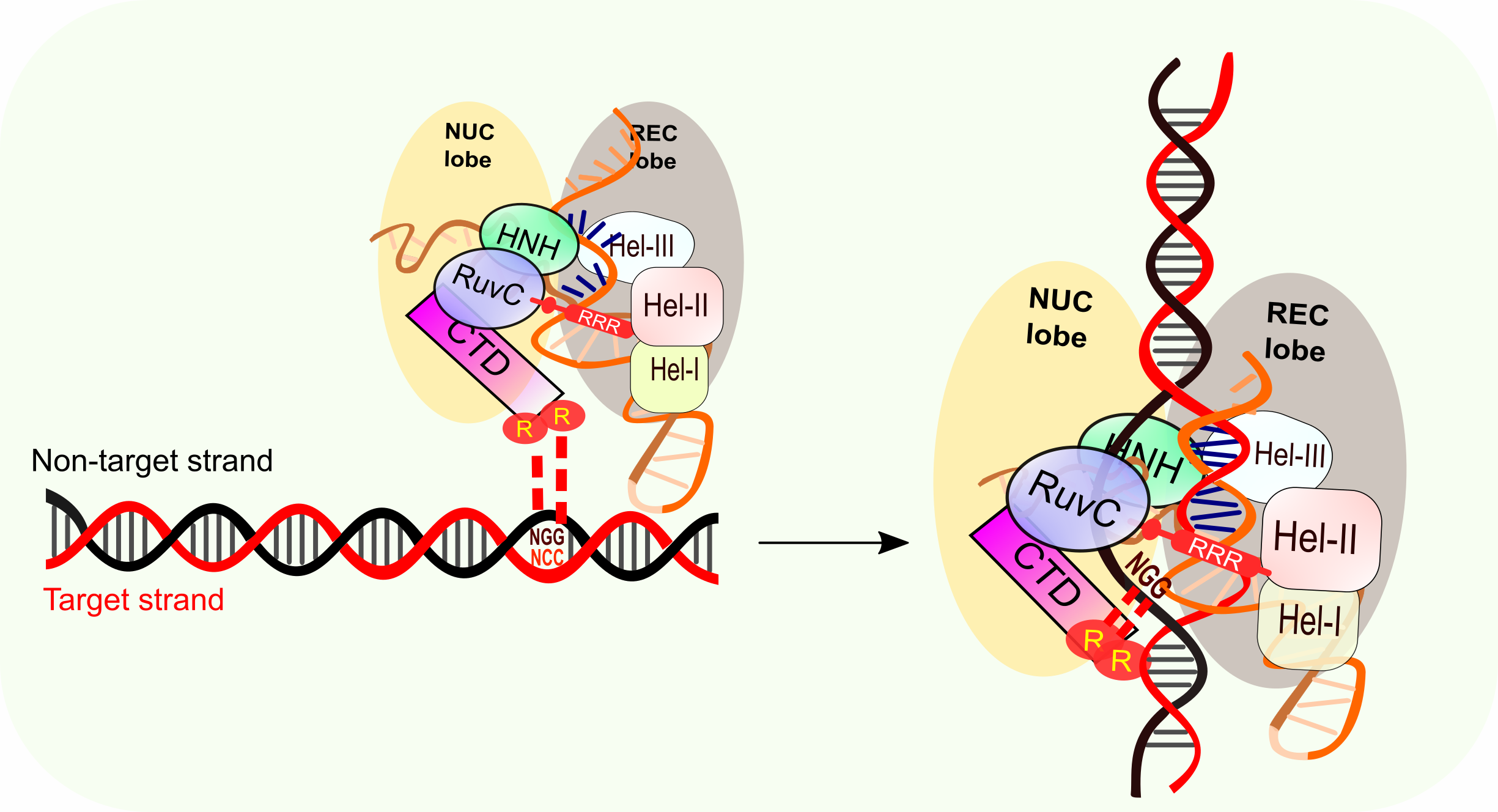 Figure 2: PAM-binding of Cas9/gRNA complex and initiation of crRNA/target DNA hybridization.
Figure 2: PAM-binding of Cas9/gRNA complex and initiation of crRNA/target DNA hybridization.
Upon PAM recognition and DNA/RNA duplex formation, each DNA strand is positioned to be cleaved by the nuclease domains HNH (target strand) and RuvC (non-target strand) 3nt upstream of the PAM. Off-target analysis suggested that there is far more Cas9/gRNA-DNA-binding events than the cleavage in the cell, which indicates that the PAM-binding alone is not enough to trigger the nuclease activity. In fact, 10-14nt complementary crRNA to the target strand is essential for the activation of the nuclease activity.
FRET experiments revealed that the distal complementary crRNA/target DNA duplex is a checkpoint which triggers a large conformational change to activate the HNH nuclease activity (Figure 3). The two hinge regions that link HNH and RuvC are important for RuvC nuclease activation to cut the non-target 3nt upstream of the NGG. Interestingly, the conformational change where HNH nuclease domain interacts with Helix-II (Hel-II) upon dsDNA binding might play important roles in locking the HNH domain in an active state. After the cleavage, the Cas9 protein remains to bind with PAM, and the in vivo mechanism to remove Cas9 to facilitate further DNA repair remains unclear.
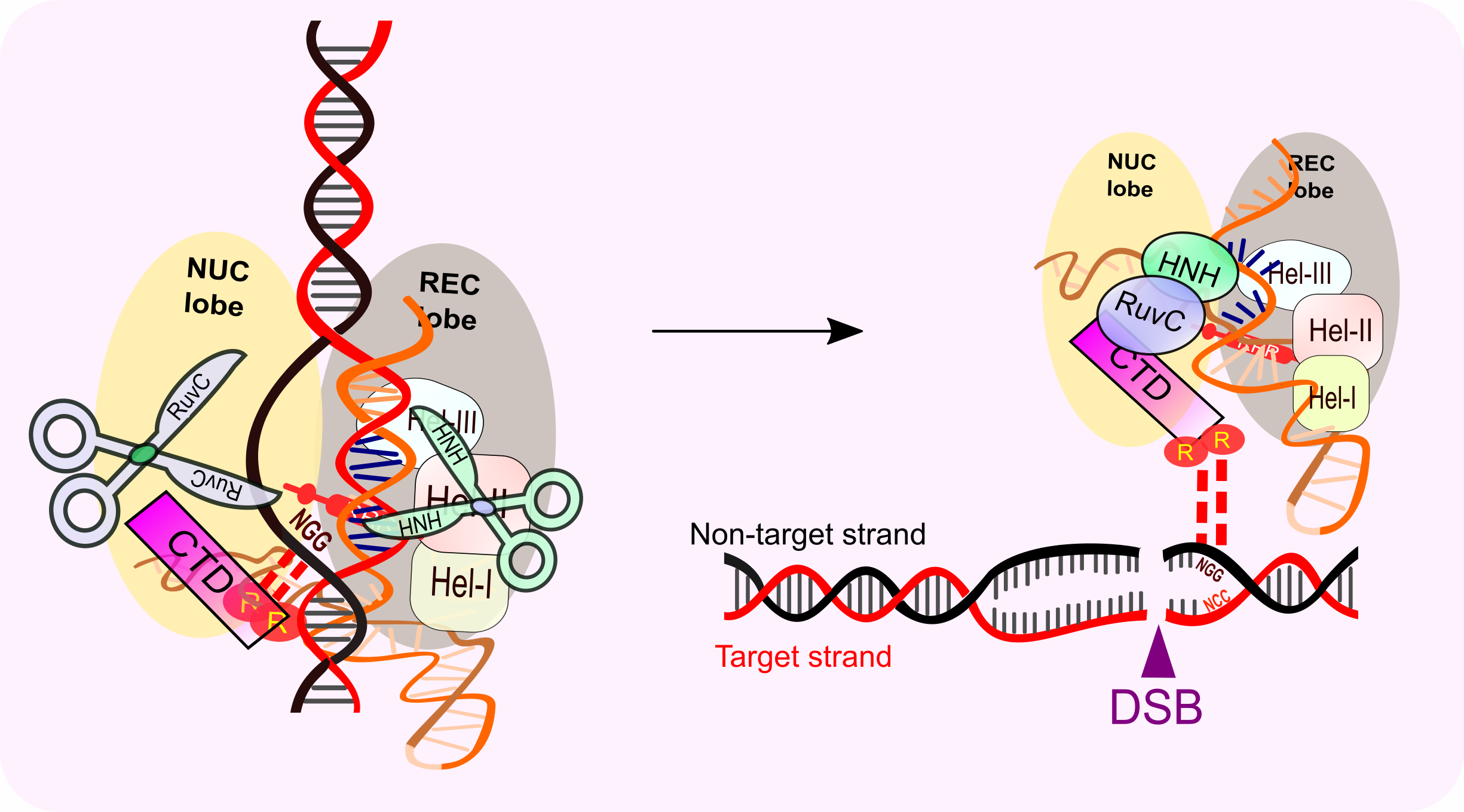 Figure 3: Activation of Cas9 HNH and Ruv-C nuclease activity to create the double strand break
Figure 3: Activation of Cas9 HNH and Ruv-C nuclease activity to create the double strand break
How can the practitioner increase efficiency decrease off-target effects?
With the structural knowledge of how CRISPR/Cas9 produces a location-specific DSB, people can control the process by protein engineering for better gene-editing or even broader applications. For example, point mutation on the nuclease domain HNH (H840A) and RuvC (D10A) converts the enzyme into nickase, which only creates a single strand break on non-target strand or the target stand, respectively. The advantage with a DNA nickase is DNA nicks tend to induce HR DNA repair pathway over the error prone NHEJ (Maizels and Davis, 2018). Interestingly, the cleavage efficiency of the paired engineered Cas9 (D10A) nickases is more efficient or comparable to individual Cas9, suggesting engineering Cas9 for specific functions is possible without loss of efficiency (Gopalappa et al., 2018). A recent study showed that fusion of Rad51 (a DNA repair protein) or its variants with Cas9(D10A) nickase promotes HR DNA-repair at the DNA nicks and decreased off-target effects in some cell lines (Rees et. al. 2019). Fusion of Cas9 with a structurally unstable protein domain such as dihydrofolate reductase (DHFR) lead to the degradation of the protein under normal conditions. With trimethoprim (TMP) application, DHFR-Cas9 can be stabilized in a short period of time for gene editing, and the TMP can be removed to limit Cas9 activity to reduce off-target effects.
What else can we do with CRISPR/Cas9?
The double mutation of H840A and D10A results in a catalytic dead enzyme dCas9, which still retains the target DNA binding ability. dCas9 is widely used in different applications by fusion with various active domains. When fused with cytidine deaminase, Cas9 can become a base editor; when fused with transcriptional activators and repressors, Cas9 can be used to specifically regulate gene expression. Fusion of cytosine DNA methyltransferase with dCas9 can confer the protein epigenetic regulation ability in a sequence specific manner. Also, fusion of a fluorescent protein with dCas9 has been used for imaging genomic loci in live cells.
The last decade has witnessed the discovery and improvement of CRISPR/Cas9 tool kit for both gene editing and other amazing applications. Yet, there still remains plenty of room for engineering more efficiency and precision to CRISPR/Cas9 function. It will be exciting to see how lessons and techniques learned in the CRISPR/Cas9 community lead to the next big thing in gene-editing. Let’s keep our eyes open to see what is going to come with the CRISPR/Cas9 legend!
References:
Jiang, Fuguo, and Jennifer A. Doudna. “CRISPR–Cas9 structures and mechanisms.” Annual review of biophysics 46 (2017): 505-529.
Gopalappa, Ramu, Bharathi Suresh, Suresh Ramakrishna, and Hyongbum Kim. “Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption.” Nucleic acids research 46, no. 12 (2018): e71-e71.
Gong, Shanzhong, Helen Hong Yu, Kenneth A. Johnson, and David W. Taylor. “DNA unwinding is the primary determinant of CRISPR-Cas9 activity.” Cell reports 22, no. 2 (2018): 359-371.
Ribeiro, Lucas F., Liliane FC Ribeiro, Matheus Q. Barreto, and Richard J. Ward. “Protein engineering strategies to expand CRISPR-Cas9 applications.” International journal of genomics 2018 (2018).
Maizels, Nancy, and Luther Davis. “Initiation of homologous recombination at DNA nicks.” Nucleic acids research 46, no. 14 (2018): 6962-6973.
Rees, Holly A., Wei-Hsi Yeh, and David R. Liu. “Development of hRad51–Cas9 nickase fusions that mediate HDR without double-stranded breaks.” Nature communications 10, no. 1 (2019): 2212.













 On our day of meetings, the
On our day of meetings, the  Reach Out:
Reach Out: